Launching a processing¶
Commands¶
The main file is located in workflows and is called segment_skull.py and should be called like a python script:
$ python workflows/segment_skull.py
N.B. if you have installed the pypi version (e.g. using pip install macapype) or a docker/singularity version, you can replace the previous command by the following command:
$ segment_skull
For container (docker and singularity), here are some examples - add your proper bindings:
$ docker run -B binding_to_host:binding_guest macatools/skullto3d:latest segment_skull
$ singularity run -v binding_to_host:binding_guest /path/to/containers/skullto3d_v0.0.4.1.sif segment_skull
Expected input data¶
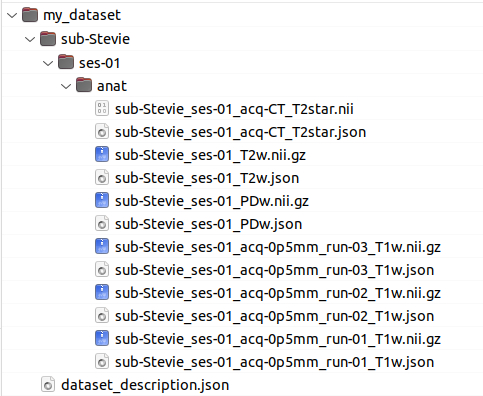
All the data have to be in BIDS format to run properly (see BIDS specification for more details)
In particular:
_T1w (BIDS) extension is expected for T1 weighted images (BIDS)
_T2w (BIDS) extension is expected for T2 weighted images (BIDS)
_PDw (BIDS) or petra (non-BIDS) extensions are expected for petra images
Note : All files with the same extension (T1w, T2w or PDw) will be aligned to the first one and averaged
_acq-CT_T2star (BIDS, but non canonical) extension is expected for CT images
_angio extension is expected for angiography images
Command line parameters¶
All parameters available for macapype are also available in skullTo3d. These parameters are recalled here
mandatory parameters¶
-data: path to your data dataset (existing BIDS format directory)-out: path to the output results (an existing path)-soft: can be one of these : SPM or ANTS ( NB: SPM requires a specific version of macapype/skullTo3d, not available by default)For
-softvalue, it is possible to add some key words (e.g.-soft ANTS_robustreg_prep) all these options are available (to place after SPM or ANTS, e.g) and will change the brain extraction:_4animal: will use bet4animal (FSL) for brain extraction, for faster computation (by default atlas_brex is used)_quick: will use hd-bet (Deep Learning) for brain extraction, for faster computation (by default atlas_brex is used) NB: hd-bet requires a specific version of macapype/skullTo3d, not available by default_robustreg(at the end) to have a more robust registration (in two steps) . This option should be used if the coregistration to template in preparation is not performed correctly
Finally, these option are available (to place after SPM or ANTS) and will modify the parameters but can be launched in sequence:
_test: (at the end) to check if the full pipeline is coherent (will only generate the graph.dot and graph.png)_prep(at the end) will perform data preparation (no brain extraction and segmentation)_noseg(at the end) will perform data preparation and brain extraction (no segmentation)_skullafter SPM or ANTS if you want to process skull or angio specific to skullTo3d; otherwise the main pipelines of macapype will be launched (only brain segmentation will be performed) NB:_skullnoisypetrainstead of_skullavailable for macaque with issues on petra NB:-soft skullwithout processing brain is possible, but is still experimental. It only works if-skull_dt CT petraand -deriv -padback are NOT defined_noheadmask(at the end) will perform only realignement to stereo_noskullmask(at the end) will perform realignement to stereo and compute headmask (only realignement for CT)_nofullskullmask(at the end) will not perform fullskullmask (only realignement for CT)
exclusive parameters¶
(but one is mandatory)
-params: (mandatory if -species is omitted) a json file specifiying the global parameters of the analysis. See Parameters for more details-species: (mandatory if -params is omitted) followed the NHP species corresponding to the image, e.g. {macaque | marmo }
NB: marmoT2 can be used for segmenting from the T2w image (by default, T1w is used for marmo) NB: macaque_0p5 is available to use downsampled template (faster results)
optional parameters¶
(but highly recommanded)
-brain_dtequivalent to -dt in macapype
specifies the datatype available to perform brain segmentation (can be “T1”, or “T1 T2”).
NB : default is T1 if the attribute is omitted
-skull_dtspecific to skullTo3d
specifies the datatype available for skull segmentation (can be, “T1”, “petra”, “CT”, “angio” or a combination of the latter (with space(s) in between).
NB : default is T1 if the attribute is omitted.
-deriv: creates a derivatives directory, with all important files, properly named following BIDS derivatives convertion. See Derivatives for a descrition of the outputs-padback: exports most important files in native (original) space
more optional parameters¶
-indivor-indiv_params: a json file overwriting the default parameters (both macapype default and parameters specified in -params json file) for specific subjects/sessions. See Individual Parameters for more details-sub(-subjects),-ses(-sessions),-acq(-acquisions),-rec(-reconstructions) allows to specifiy a subset of the BIDS dataset respectively to a range of subjects, session, acquision types and reconstruction types. The arguments can be listed with space seperator. Note if not specified, the full BIDS dataset will be processed-nprocs: an integer, to specifiy the number of processes that should be allocated by the parralel engine of macapypetypically equals to the number of subjects*session (i.e. iterables).
can be multiplied by 2 if T1*T2 pipelines are run (the first steps at least will benefit from it)
default = 4 if unspecified ; if is put to 1, then the sequential processing is used
-maskallows to specify a precomputed binary mask file (skipping brain extraction). The best usage of this option is: precomputing the pipeline till brain_extraction_pipe, modify by hand the mask and use the mask for segmentation. Better if only one subject*session is specified (one file is specified at a time…).
Warning: the mask should be in the same space as the data. And only works with -soft ANTS so far
The following parameters are optional¶
-indiv or -indiv_params : a json file overwriting the default parameters (both macapype default and parameters specified in -params json file) for specific subjects/sessions. See Individual Parameters for more details
-sub (-subjects), -ses (-sessions), -acq (-acquisions), -rec (-reconstructions) allows to specifiy a subset of the BIDS dataset respectively to a range of subjects, session, acquision types and reconstruction types. The arguments can be listed with space seperator. Note if not specified, the full BIDS dataset will be processed
-mask allows to specify a precomputed binary mask file (skipping brain extraction). The best usage of this option is: precomputing the pipeline till brain_extraction_pipe, modify by hand the mask and use the mask for segmentation. Better if only one subject*session is specified (one file is specified at a time…).
Warning: the mask should be in the same space as the data. And only works with -soft ANTS so far
- -nprocsan integer, to specifiy the number of processes that should be allocated by the parralel engine of macapype
typically equals to the number of subjects*session (i.e. iterables).
can be multiplied by 2 if T1*T2 pipelines are run (the first steps at least will benefit from it)
default = 4 if unspecified ; if is put to 0, then the sequential processing is used (equivalent to -soft with _seq, see before)
Command line examples¶
$ python workflows/segment_skull.py -data ~/Data_maca -out ./local_test -soft ANTS_skull -params params.json
$ python workflows/segment_skull.py -data ~/Data_maca -out ./local_test -soft ANTS_skull_robustreg -species macaque
$ python workflows/segment_skull.py -data ~/Data_maca -out ./local_test -soft ANTS_skull -params params.json -sub Apache Baron -ses 01 -rec mean -deriv -padback